
材料的性质由其原子排列决定。然而,现有的获得这种排列的方法要么过于昂贵,要么对许多元素无效。
现在,加州大学圣地亚哥分校纳米工程系的研究人员开发了一种人工智能算法,可以几乎即时地预测任何材料(无论是现有材料还是新材料)的结构和动态特性。该算法被称为 M3GNet,用于开发 matterverse.ai 数据库,该数据库包含超过 3100 万种尚未合成的材料,其特性由机器学习算法预测。Matterverse.ai 促进了具有卓越性能的新技术材料的发现。
该研究以「A universal graph deep learning interatomic potential for the periodic table」为题,于 2022 年 11 月 28 日发布在《Nature Computational Science》上。

论文链接:https://www.nature.com/articles/s43588-022-00349-3
对于大规模材料研究,需要根据原子之间的多体相互作用来描述势能面 (PES) 的高效、线性标度的原子间势 (IAP)。然而,如今大多数 IAP 都是为范围很窄的化学物质定制的:通常是单一元素或最多不超过四到五种元素。
最近,PES 的机器学习已成为一种特别有前途的 IAP 开发方法。然而,还没有研究证明在元素周期表和所有类型的晶体中有一个普遍适用的 IAP。
在过去的十年中,高效、可靠的电子结构代码和高通量自动化框架的出现,导致了计算材料数据的大型联邦数据库的发展。在进行结构松弛过程中积累了大量的 PES 数据,即中间结构及其相应的能量、力和应力,但人们对这些数据关注较少。
「与蛋白质类似,我们需要了解材料的结构才能预测其特性。」 该研究的主要作者 Shyue Ping Ong说。「我们需要的是用于材料的 AlphaFold。」
AlphaFold 是谷歌 DeepMind 开发的一种预测蛋白质结构的人工智能算法。为了构建材料的等价物,Ong 和他的团队将图神经网络与多体交互(many-body interactions)相结合,构建了一个深度学习架构,该架构可以在元素周期表的所有元素中通用、高精度地工作。
数学图是晶体和分子的自然表示,节点和边分别代表原子和它们之间的键。传统的材料图神经网络模型已被证明对一般材料特性预测非常有效,但由于缺乏物理约束,因此不适合用作 IAP。
研究人员开发了一个明确包含多体相互作用的材料图架构。模型开发的灵感来自传统的 IAP,在这项工作中,将重点关注三体交互 (M3GNet) 的整合。
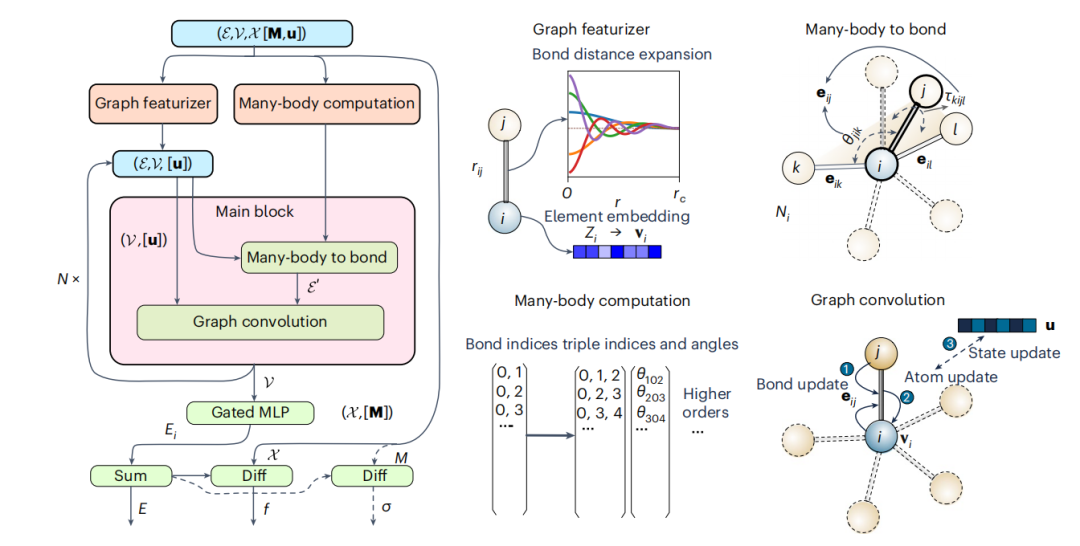
图 1:多体图势和主要的计算块示意图。(来源:论文)
IAP 数据集的基准测试
作为初始基准,研究人员择了 Ong 和同事先前生成的元素能量和力的多样化 DFT 数据集,用于面心立方(fcc)镍、fcc 铜、体心立方(bcc)锂、bcc 钼、金刚石硅和金刚石锗。
表 1:M3GNet 模型与现有模型 EAM、MEAM、NNP 和 MTP 在单元素数据集上的误差比较。(来源:论文)
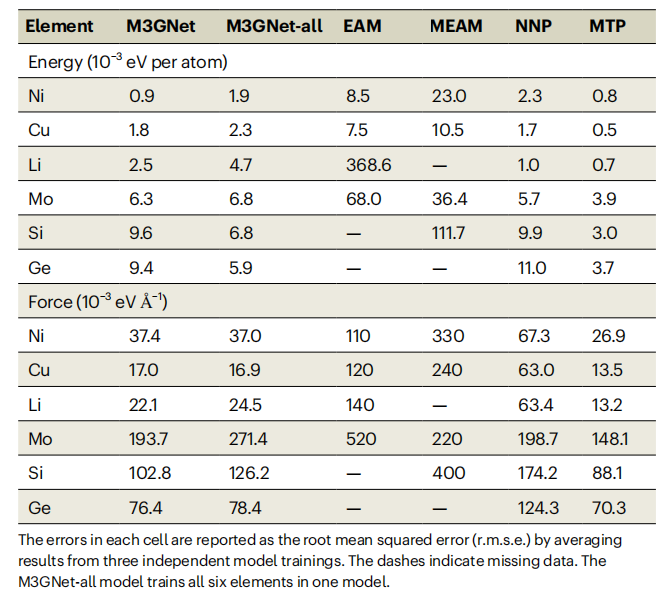
从表 1 可以看出,M3GNet IAP 大大优于经典的多体势;它们的性能也与基于本地环境的 ML-IAP 相当。应该注意的是,尽管 ML-IAP 可以实现比 M3GNet IAP 略小的能量和力误差,但它在处理多元素化学方面的灵活性会大大降低,因为在 ML-IAP 中加入多种元素通常会导致组合爆炸回归系数的数量和相应的数据要求。相比之下,M3GNet 架构将每个原子(节点)的元素信息表示为可学习的嵌入向量。这样的框架很容易扩展到多组分化学。
与其他 GNN 一样,M3GNet 框架能够捕获长距离的相互作用,而无需增加键构建的截止半径。同时,与之前的 GNN 模型不同,M3GNet 架构仍然随着键数的变化保持能量、力和应力的连续变化,这是 IAP 的关键要求。
元素周期表的通用 IAP
为了开发整个元素周期表的 IAP,该团队使用了世界上最大的 DFT 晶体结构弛豫开放数据库之一(Materials Project)。
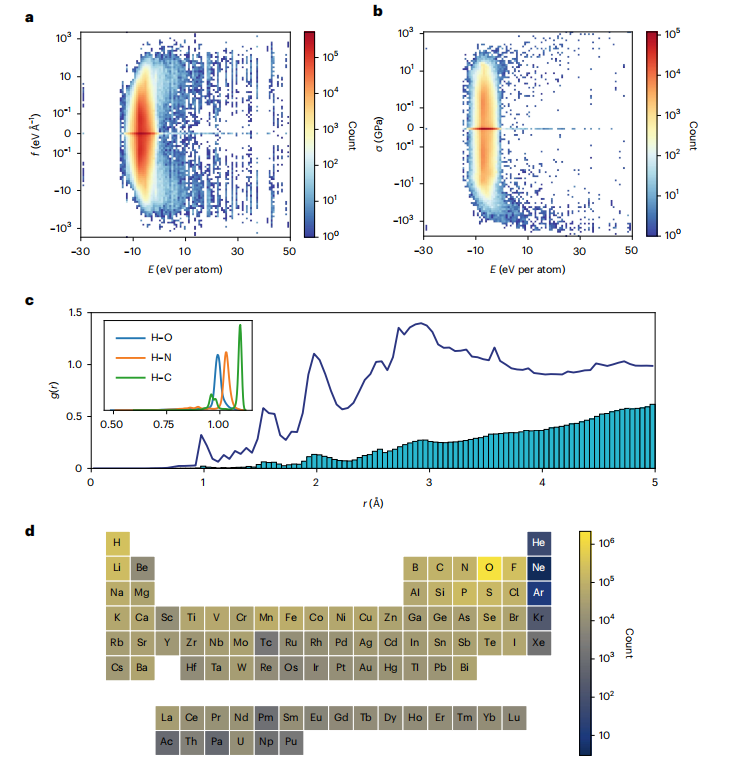
图 2:MPF.2021.2.8 数据集的分布。(来源:论文)
原则上,IAP 可以只训练能量,或者能量和力的组合。在实践中,仅在能量上训练的 M3GNet IAP (M3GNet-E) 无法达到预测力或应力的合理精度,平均绝对误差 (MAE) 甚至大于数据的平均绝对偏差。能量+力(M3GNet- EF)和能量+力+应力(M3GNet-EFS)训练的 M3GNet 模型获得了相对相似的能量和力 MAE,但 M3GNet- EFS 的应力 MAE 约为 M3GNet- EF 模型的一半。
对于涉及晶格变化的应用,例如结构松弛或 NpT 分子动力学模拟,准确的应力预测是必要的。研究结果表明,在模型训练中包含所有三个属性(能量、力和压力)对于获得实用的 IAP 至关重要。最终的 M3GNet-EFS IAP(以下简称为 M3GNet 模型)实现了每个原子 0.035eV 的平均值,能量、力和压力测试 MAE 的平均值分别为 0.072eVÅ−1 和 0.41GPa。
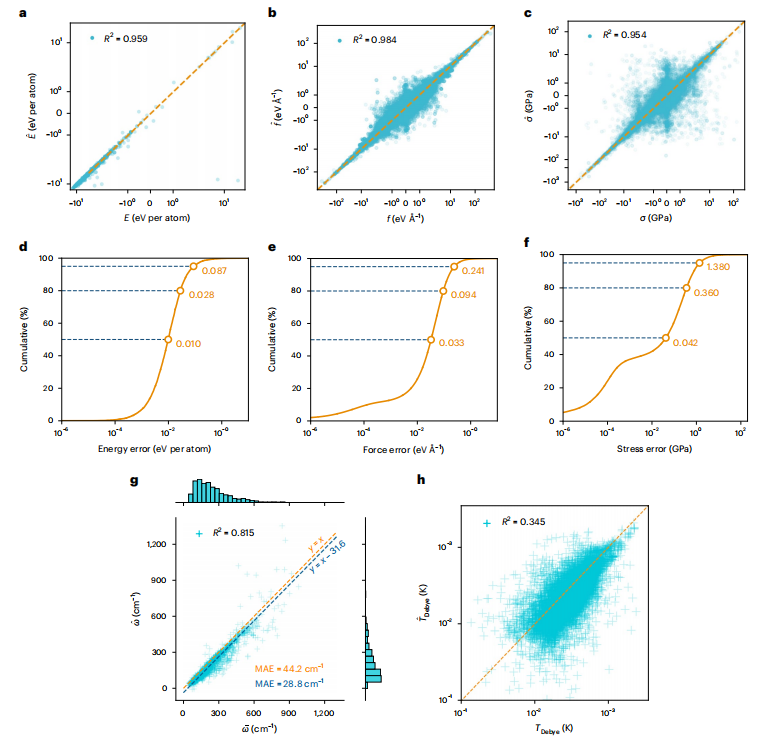
图 3:与 DFT 计算相比,测试数据集上的模型预测。
在测试数据上,模型预测和 DFT ground truth 匹配得很好,正如 DFT 和模型预测之间线性拟合的高线性度和 R2 值所揭示的那样。模型误差的累积分布表明,50% 的数据的能量、力和应力误差分别小于每个原子 0.01eV、0.033eVÅ−1 和 0.042 GPa。M3GNet 计算的德拜温度不太准确,这可归因于 M3GNet 对剪切模量的预测相对较差;然而,体积模量预测是合理的。
然后将 M3GNet IAP 应用于模拟材料发现工作流程,其中最终的 DFT 结构是先验未知的。M3GNet 松弛是对来自 3,140 种材料的测试数据集的初始结构进行的。M3GNet 松弛结构的能量计算产生每个原子 0.035 eV 的 MAE,并且 80% 的材料的误差小于每个原子 0.028 eV。使用 M3GNet 松弛结构的误差分布接近于所知道 DFT 最终结构的情况,这表明 M3GNet 潜力可以准确地帮助获得正确的结构。一般来说,M3GNet 的松弛会迅速收敛。
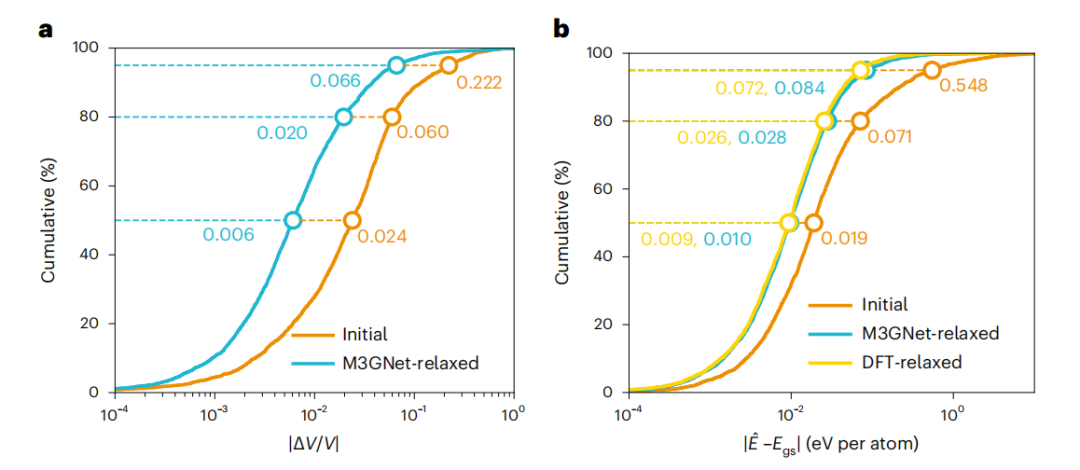
图 4:使用 M3GNet 弛豫晶体结构。(来源:论文)
新材料发现
M3GNet 能准确、快速地弛豫任意晶体结构,并预测它们的能量,使其成为大规模材料发现的理想选择。研究人员生成了 31,664,858 个候选结构作为起点,使用 M3GNet IAP 松弛结构并计算到 Materials Project 凸包 (Ehull-m) 的符号能量距离;1,849,096 种材料的 Ehull-m 每个原子小于 0.01 eV。
作为对 M3GNet 在材料发现方面性能的进一步评估,研究人员计算了发现率,即从约 180 万 Ehull-m小于 0.001 eV /原子的材料中均匀采样 1000 个结构的 DFT 稳定材料(Ehull−dft ≤ 0)的比例。发现率保持接近 1.0,达到每个原子约 0.5 eV 的 Ehull-m 阈值,并且在每个原子 0.001 eV 的最严格阈值下保持在 0.31 的合理高值。
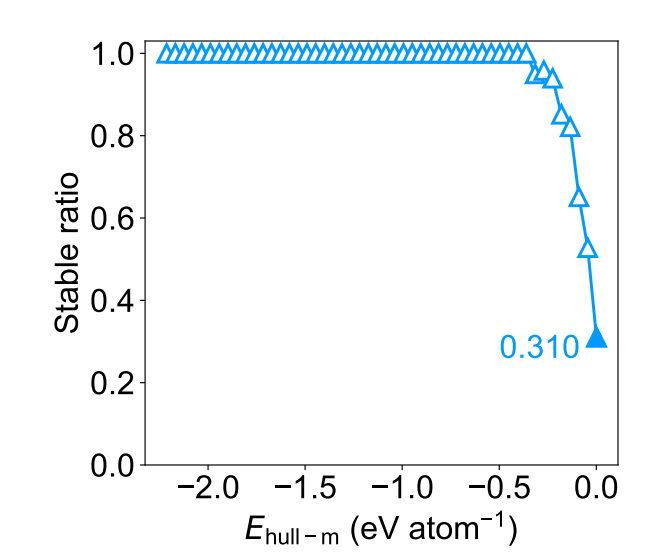
图 5:对于 1000 个结构的均匀样本,DFT 稳定比作为 Ehull−m 阈值的函数。(来源:论文)
对于这个材料集,研究人员还比较了有无 M3GNet 预松弛的 DFT 松弛时间成本。结果表明,没有M3GNe t预松弛时,DFT 松弛时间成本约为 M3GNet 预松弛时的 3 倍。
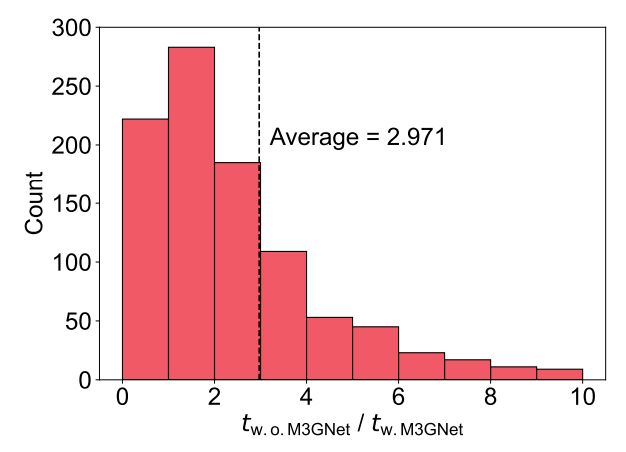
图 6:使用 M3GNet 预松弛的 DFT 加速。(来源:论文)
在今天 matterverse.ai 的 3100 万种材料中,预计有超过 100 万种材料具有潜在的稳定性。Ong 和他的团队不仅打算大大扩展材料的数量,还打算大幅扩展 ML 预测属性的数量,包括使用他们之前开发的多保真度方法的小数据量的高价值属性。
除了结构松弛,M3GNet IAP 在材料动态模拟和性能预测方面也有广泛的应用。
「例如,我们通常对锂离子在锂离子电池电极或电解质中的扩散速度很感兴趣。扩散越快,电池充电或放电的速度就越快,」Ong 说。「我们已经证明,M3GNet IAP 可用于以高精度预测材料的锂电导率。我们坚信 M3GNet 架构是一种变革性工具,可以极大地扩展我们探索新材料化学和结构的能力。」
为了推广 M3GNet 的使用,该团队已将该框架作为开源 Python 代码发布在 Github 上。并计划将 M3GNet IAP 作为工具集成到商业材料模拟包中。
© 版权声明
文章版权归作者所有,未经允许请勿转载。


