图 0
不同种类细胞中基因组构象的差异决定了基因表达的特异性,进而决定不同细胞类型的功能差异。长久以来,从原位杂交到高通量检测如 Hi-C、micro-C 技术,基因组构象检测的实验方法通常耗时耗力、成本高昂且有很强的技术局限性。这些方法极大地限制了这些实验技术在基因组构象研究领域的广泛应用,尤其是研究稀有细胞类型以及需要大规模验证基因组构象调控的因果关系等方面。这些方法的局限性也长期限制三维基因组构象调控领域里的新发现。
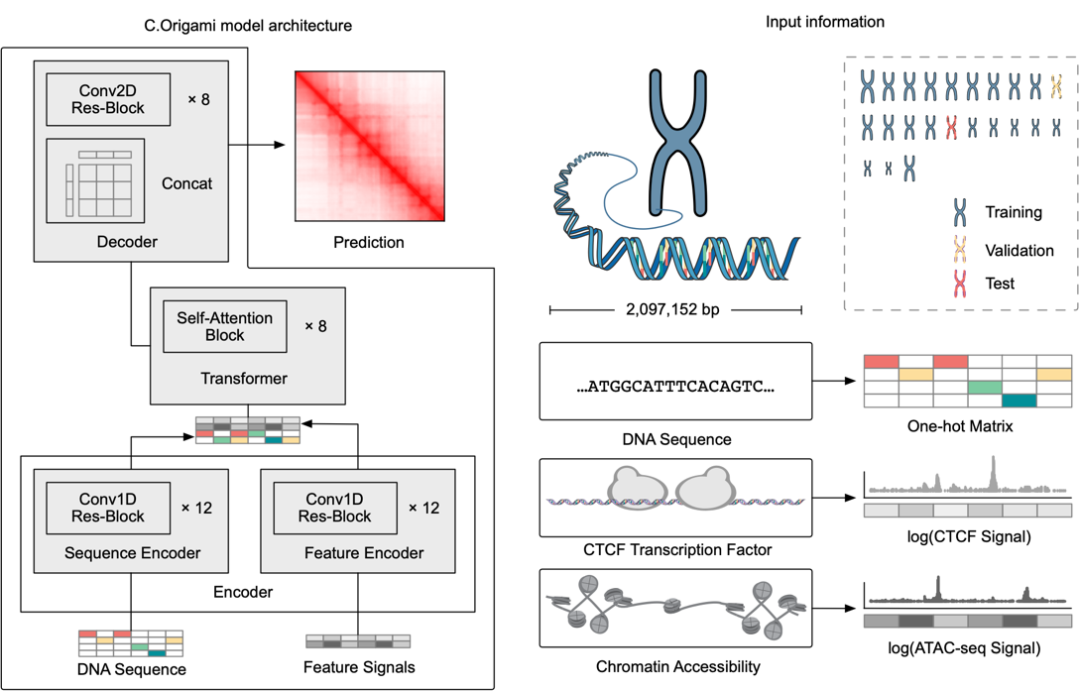
图 1
2023 年 1 月 9 日,纽约大学医学院(NYU Grossman School of Medicine)Aristotelis Tsirigos 实验室和博德研究所(Broad Institute of MIT and Harvard)夏波实验室合作在 Nature Biotechnology 上发表文章《Cell type-specific prediction of 3D chromatin organization enables high-throughput in silico genetic screening》。

论文地址:https://www.nature.com/articles/s41587-022-01612-8
这项研究中,第一作者纽约大学医学院博士生谭济民与夏波博士首先提出了新型多模态机器学习模型 C.Origami 来预测特定细胞类型的染色质构象,并基于遗传筛选的原理提出了全新的高通量计算遗传筛选 (in silico genetic screening, ISGS) 方法,用以鉴定细胞类型特异性的功能基因组元件,助力发现新的染色质构象调控机理。
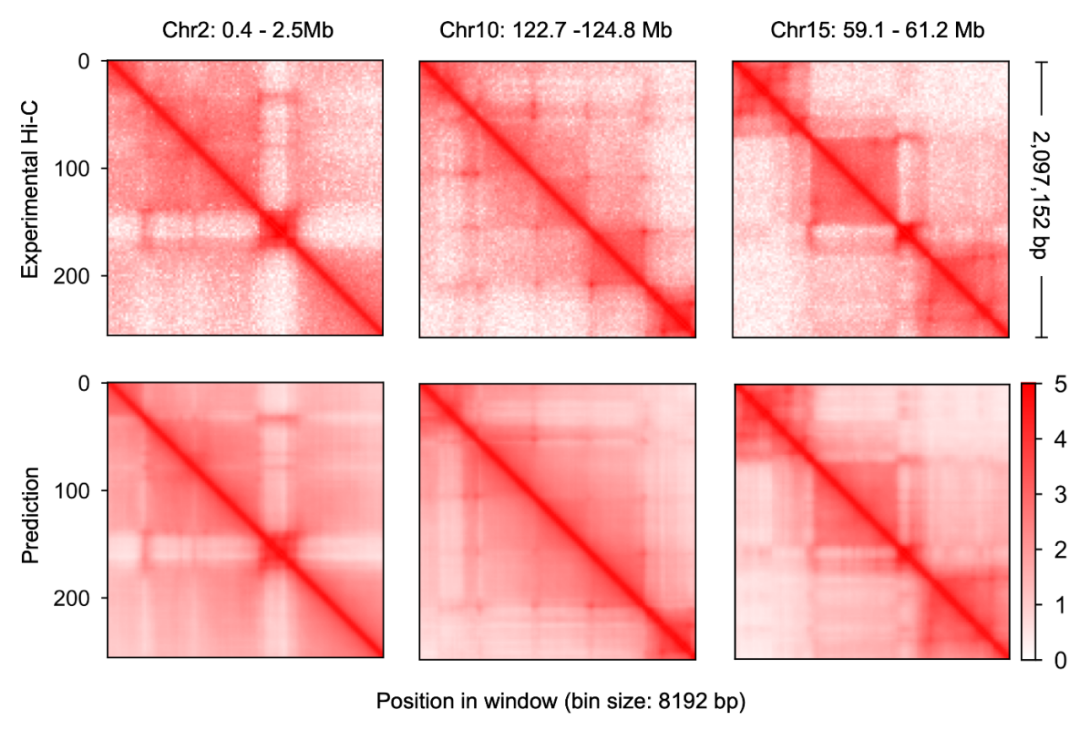
图 2
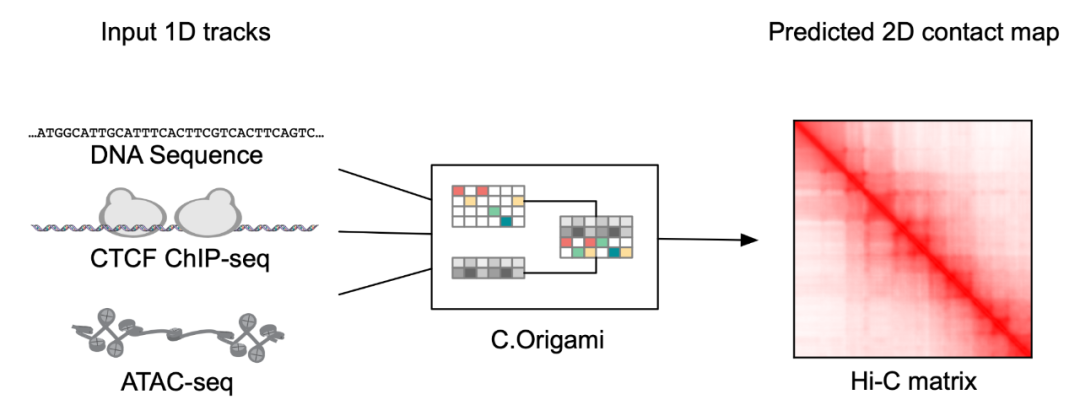
研究者首先构建了应用于基因组数据的新型多模态深度学习框架,Origami,使其能有效地整合 DNA 序列信息以及细胞特异性的功能基因组信息,进而预测新的基因组信息。通过反复调试及模型训练,研究者发现整合 DNA 序列、CTCF 结合状态(CTCF ChIP-seq)、及 ATAC-seq 信号作为输入信息可以准确地预测染色质构象,并以二维的 Hi-C 矩阵作为预测输出目标(图 1-2)。输入信息为 2 百万碱基对的 DNA,CTCF ChIP-seq 和 ATAC-seq。研究者们使用 Onehot-encoding 来编码离散的 DNA 序列,而 CTCF ChIP-seq 和 ATAC-seq 则编码成非离散的特征。
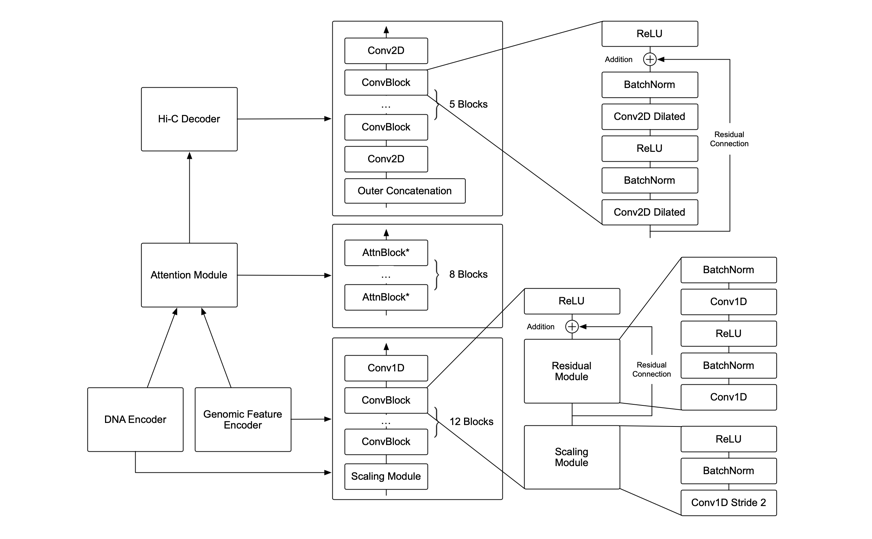
C.Origami 模型分为三个部分,处理并压缩 DNA 及基因组信息的编码器,Transformer 中间层和输出 Hi-C 解码器。其中编码器由一系列 1D ResNet 和 strided convolution 构成用来编码和压缩 2 百万碱基对的输入信息。在编码器末端 2 百万长度的信息被压缩为 256 长度并作为 Transformer 的输入信息。Transformer 的自注意力机制可以处理不同基因组区域间的 interdependency 并提升了模型的综合性能。Transformer 中的注意力矩阵还可以增强模型的可解释性。研究者们将注意力权重转换成了 “attention score ”,用来衡量模型在预测时对于不同区域的侧重。最后,研究者们将 Transformer 模块的 1D 输出用 “outer concatenation” 的方式转换成了 2D 的 contact/adjacency matrix,用作 Hi-C 解码器的输入信息。解码器是一个 Dilated 2D ResNet。研究者们调整了不同层的 dilation factor 使得最后层的每一个像素位置的 receptive field 都能覆盖所有输入信息。
这一预测染色质构象的模型则被称为 C.Origami。研究者称 C.Origami 是基因组学中第一个多模态深度学习模型。由于它多模态的特性,C.Origami 能够准确地预测(de novo prediction)从未接触过的新细胞类型的染色质构象。例如,在 IMR-90 细胞(肺成纤维细胞)上训练的模型能够准确预测出 GM12878 细胞(B 淋巴细胞)里特定的染色质构象(图 3)。
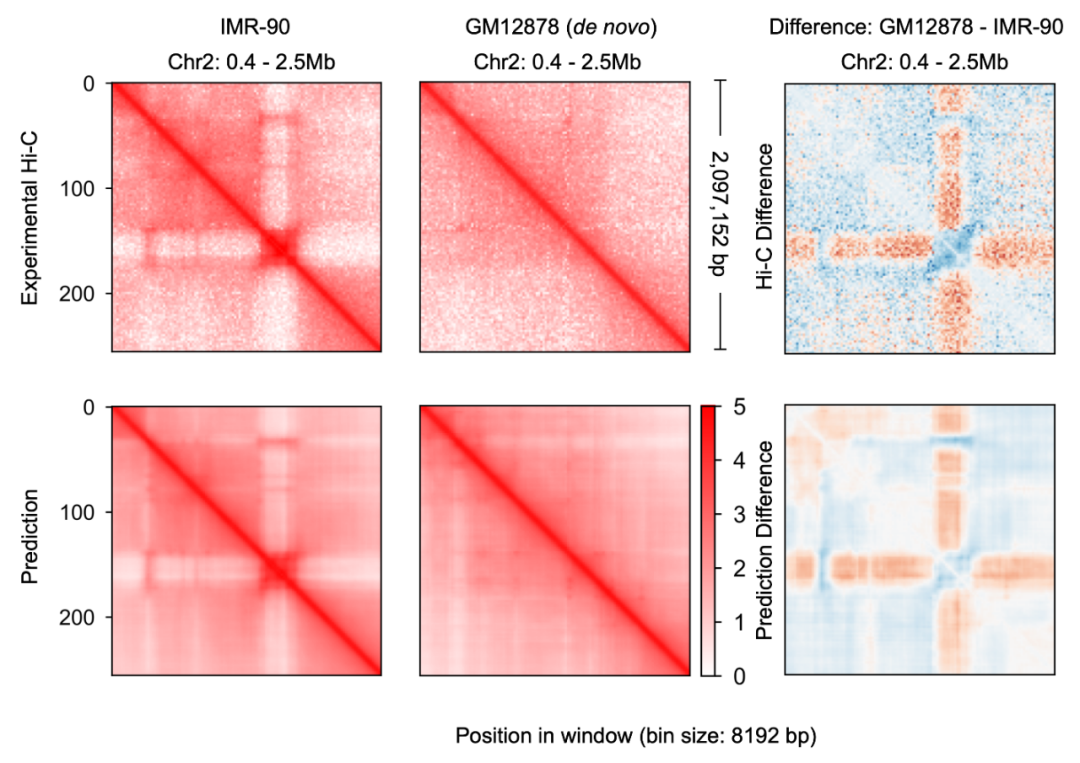
图 3
结构变异(structural variant)—- 比如染色体易位 —- 在肿瘤中非常常见,并经常改变染色质相互作用模式,进而可能影响癌基因或抑癌基因的表达。研究这些结构变异对染色质构象及基因表达的影响对理解肿瘤发生和进展的机理有重要作用。这类研究通常需要借助 4C-seq 或 Hi-C 等实验来分析结构变异位点的染色质构象,但又往往受限于资源和时间的限制,难以大规模开展。
这项研究中,C.Origami 可以在输入变量中模拟 DNA 序列的变异,然后预测变异后的癌症基因组中新的染色质相互作用。之前的研究发现 T 细胞急性淋巴细胞白血病(T-ALL)细胞模型 CUTLL1 有一个 chr7-chr9 的染色体易位(图 4)。通过计算模拟染色体易位变异,C.Origami 准确预测了在变异位点的新 TAD 结构,并检测到从 chr9 延伸到 chr7 的‘互动束’(chromatin stripe)结构(图 4)。
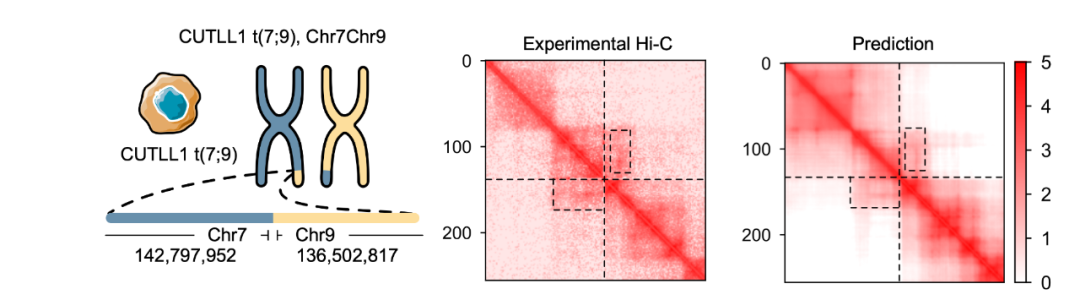
图 4
鉴于 C.Origami 的精准预测效果,并受到反向遗传筛选原理的启发,研究者提出了全新的高通量计算遗传筛选 (in silico genetic screening,ISGS) 方法,用以系统鉴定细胞类型特异性的功能基因组元件,并助力发现新的染色调控分子(图 5)。研究者们基于 C.Origami 模型开发了用于系统性鉴定染色质构象所需的顺式调控元件(cis-regulatory element)的计算遗传筛选 ISGS 的框架。通过对全基因组 1kb 分辨率的 ISGS,作者分离出对染色质构象有重要影响的顺式调控元件(占约 1% 的基因组)。这些染色质构象调控序列呈现出对 CTCF 结合和 ATAC-seq 信号的不同依赖度(图 5)。
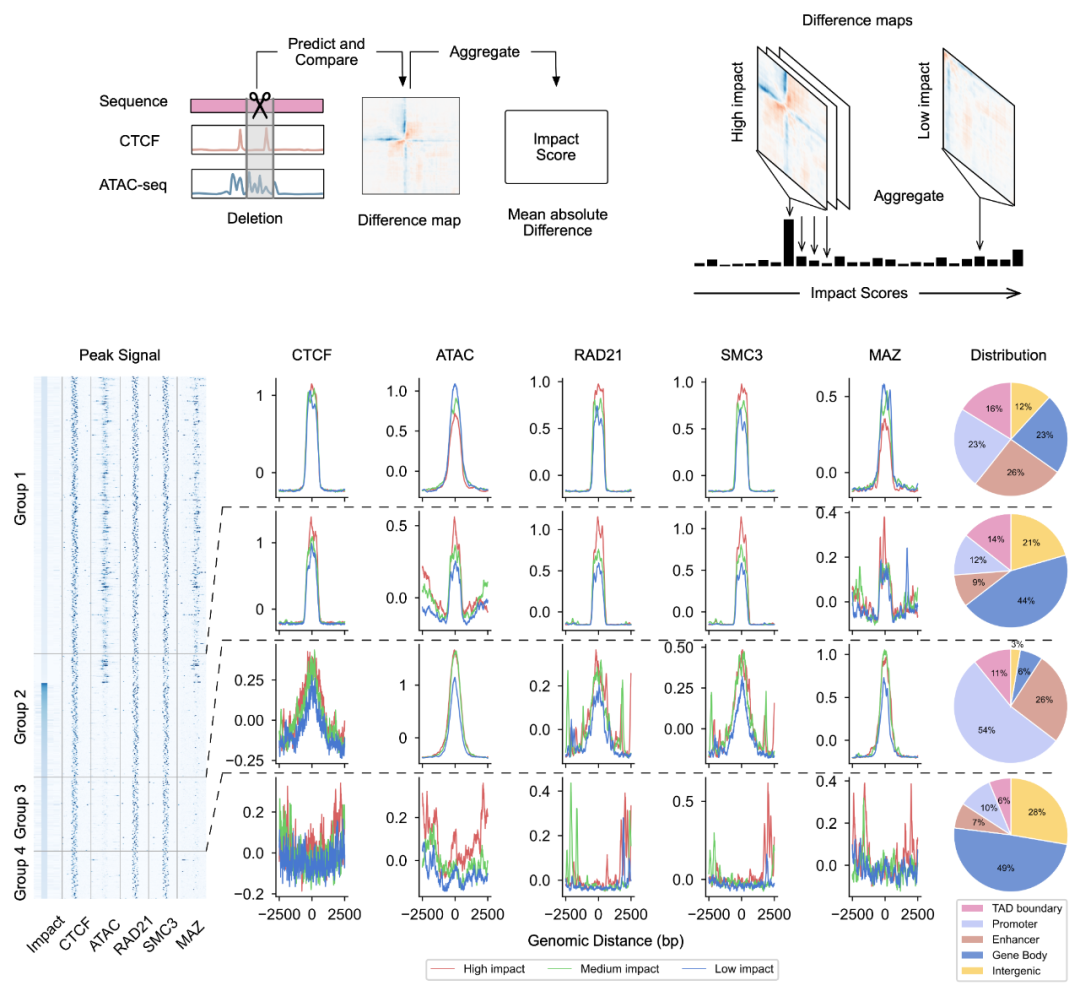
图 5
ISGS 框架可以对细胞或疾病特异性的染色质构象进行高通量筛选。研究者在 CUTLL1、Jurkat 和正常 T 细胞中分别进行了 ISGS,并发现了一个在 CHD4 基因附近的顺式调控元件 (CHD4-insu) 在 T-ALL 细胞里特异性丢失。筛选结果表明,T-ALL 细胞中 CHD4-insu 的绝缘性缺失可能使 CHD4 基因建立新的染色质相互作用,进而上调 CHD4 表达并促进白血病细胞增殖。
ISGS 也可以用来系统地发现调控染色质构象的新型反式作用因子(trans-acting factors)。通过对细胞类型特异性的重要调控序列与转录因子结合部位的富集分析,研究者确定了有助于细胞类型特异的基因组构象的调控因子。有意思的是,之前研究发现 MAZ 可能与 CTCF 一起调控染色质构象。通过 ISGS 及转录因子富集分析,作者发现 MAZ 极大地富集于开放染色质区域,而在 CTCF 结合的非开放染色质区域仅显示微弱结合。这一结果预示 MAZ 可能独立于 CTCF 调节基因组构象。
研究者们在染色质结构预测中看到了结合 DNA 序列与染色质信息的多模态机器学习模型的巨大潜力。而该模型的底层多模态架构 Origami 可以推广到其他基因组学数据的应用,例如表观遗传修饰、基因表达、突变功能性筛查等。研究者预测,未来的基因组学研究将更多的转向使用利用深度学习模型作为工具来进行主要计算遗传筛选,并辅以生物实验验证的新一代高通量研究方法。
这项研究中,纽约大学医学院博士研究生谭济民为第一作者,Aristotelis Tsirigos 博士和夏波博士为共同通讯作者。这项研究起始于 2020 年 10 月疫情封控期间夏波与谭济民的头脑风暴,历经两年半的完善与打磨,在 2023 年一月于 Nature Biotechnology 正式刊出。
此项目的代码和训练数据已在GitHub和Zenodo上开源,并配有Google Colab做功能展示。
项目地址:https://github.com/tanjimin/C.Origami
通讯作者
夏波博士实验室(Broad Institute of MIT and Harvard)主页:www.boxialab.org
夏波博士致力于解析调控基因组三维构象的核心机理及其对人类疾病、发育及进化的生物学意义。夏波实验室欢迎志同道合的博士后加入团队。
Tsirigos Lab(New York University Grossman School of Medicine)主页:http://www.tsirigos.com
Tsirigos Lab 的主要研究方向包括染色质,表观遗传学和机器学习在精准医疗中的应用。
© 版权声明
文章版权归作者所有,未经允许请勿转载。